
Gromacs is used to perform molecular dynamics simulations. It's a popular tool and we decided to install it on our cluster based on the needs of two researchers, one at UCT and the other in Kenya. Gromacs is designed to work on many platforms from desktops to high end clustered systems. It is optimized to run in multi threaded systems as well as over multiple nodes via MPI.
There are a number of different ways to install the software. As we are running Scientific Linux we decided to install via the EPEL repository rather than download an RPM or compile from scratch. While the instructions on the Gromacs site claim that installation is simple, in order to get the package to interact with FFT, as well as compile with double precision support, multi thread and MPI capability is not trivial. Gromacs' simple installation instructions can be found here and we decided that while making for an interesting read we'd rather not grapple with them. So a quick "yum install gromacs" was all that was required. Right?
No, not quite so simple. The EPEL repo has a separate install for MPI, so another "yum install gromacs-openmpi" was required. Then it was a matter of discovering that under the new EPEL compile of Gromacs 4.5.3 the binaries have all been renamed and now start with "g_" so mdrun becomes g_mdrun. However once that hurdle was overcome running the software was fairly simple. Right?
Well, not quite. While the program ran (and here one of our researchers at UCT was very helpful in providing some test cases), the software seemed to perform a bit, well, greedily. Running on a single worker node, or even multiple worker nodes it grabbed as many cores as were available, which is not great for the scheduling software as it looses track of how many cores are free for other applications. Some research indicated that mdrun no longer needed its own -np argument as Gromacs version 4 and above makes use of mpirun's -np argument. However it was only after we introduced the -nt argument for mdrun that we finally regained control over the number of cores used. Problem solved. Right?
Not really. After examining the output our UCT user noticed that multiple output files were being created, almost as if independent instances of the software was being run on each node. After some investigation it turned out that this was exactly what was happening, and then it dawned on us that this was why the software had not behaved properly in the first instance and required the -nt argument. The problem was that we were running a non MPI aware compilation that had been configured only for multi-threading. After doing some searching through the file system we found what we were looking for, g_mdrun_openmpi, which strangely wasn't on the search path which was why we'd missed it initially. Running this with mpirun would surely solve the problem. Right?
Well, almost. Turns out that EPEL's RPM was compiled without shared library support. Trying to run the software even directly on the worker node gave the following nasty message: Error while loading shared libraries: libgmxpreprocess_openmpi.so.6: cannot open shared object file. Manually adding the library to the LD_LIBRARY environment variable fixed the problem which should allow one to submit jobs to the scheduler. Right?
Well technically yes. However the jobs still failed because of the shared library problem. Exporting the environment variable in the shell script or even placing it in the profile didn't seem to work. We're not quite sure why but it may have something to do with how mpirun connects to the worker nodes. Feeling a bit desperate now we pulled a sneaky trick and used mpirun's environment argument -x to set the LD_LIBRARY variable.
Finally success. Jobs start on multiple cores, ignore the -nt argument, and the number of threads started equals the -np argument, correctly spread over the available cores in the cluster. There is only one log file, one topology file and one energy file, as expected. The log file indicates that there are a number of nodes being used equivalent to the -np argument. Running on more nodes also seems to introduce a significant speed improvement. The only fly in the ointment seems to be the visualization files with two of them being updated simultaneously. These are meant to be used with the Grace visualization package, are not compulsory and can even be disabled.
Running a job now shows how rapidly the threads are distributed over the cores in the following image, snapshots taken 10 seconds apart.
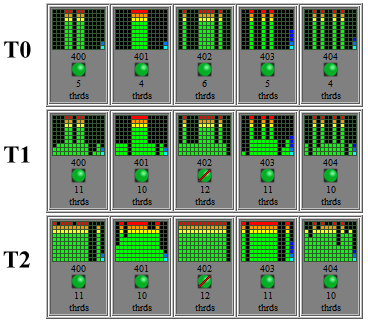 More investigation is still to be done in looking at the efficiency of multiple threads, for instance is the relationship between threads and job speed inversely geometric as expected and if so what is the minimum number of cores that will achieve at least a 90% speed improvement? The communication latency between nodes will also be examined as multiple cores seem to consume considerable bandwidth.
More investigation is still to be done in looking at the efficiency of multiple threads, for instance is the relationship between threads and job speed inversely geometric as expected and if so what is the minimum number of cores that will achieve at least a 90% speed improvement? The communication latency between nodes will also be examined as multiple cores seem to consume considerable bandwidth.
More investigation is still to be done in looking at the efficiency of multiple threads, for instance is the relationship between threads and job speed inversely geometric as expected and if so what is the minimum number of cores that will achieve at least a 90% speed improvement? The communication latency between nodes will also be examined as multiple cores seem to consume considerable bandwidth.