
Having installed Gromacs last week we decided to run some tests to see how well it performed on our cluster. We downloaded a tutorial which analysed solutions of funnel web spider venom in water. Given the bounding block size of the virtualized water space (1 angstrom around the protein) Gromacs refused to go beyond 4 simultaneous cores as it was not possible to break this job into more then four meaningful problem sets.
It quickly became clear that with large problem sets the assistance of high speed networking would assist in achieving faster computations. The communication requirement between servers over a public network is fairly high, topping out at around 5% of a GB connection. In an environment where a large number of jobs (100+) are competing for bandwidth this would introduce significant latency.
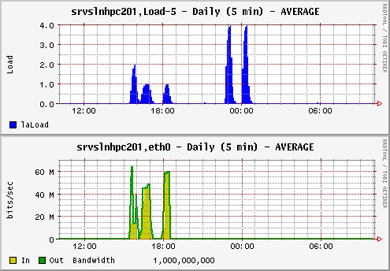 Note that the load for the parallel openMPI jobs (the first three jobs) is lower than for the OMP jobs. This is because the load for OMP is kept on one server only, while openMPI distributes the load over as many servers as the job parameters allow. The trade off however is that the network communication between the distributed processes is high. Despite this the completion times between OpenMPI and OMP were fairly similar:
Note that the load for the parallel openMPI jobs (the first three jobs) is lower than for the OMP jobs. This is because the load for OMP is kept on one server only, while openMPI distributes the load over as many servers as the job parameters allow. The trade off however is that the network communication between the distributed processes is high. Despite this the completion times between OpenMPI and OMP were fairly similar:
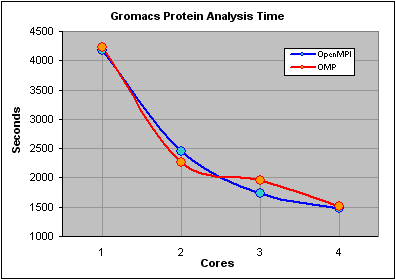 We were expecting the non-openMPI time for one core to exceed the completion time for the openMPI job "distributed" over 1 core and were surprised to see that the times were almost identical. After some head scratching we realized that this was because the non-openMPI job was still a parallel job, using OMP, as the software had been compiled with this option enabled. In fact by not specifying the -nt parameter the job automatically grabs as many free cores as possible.
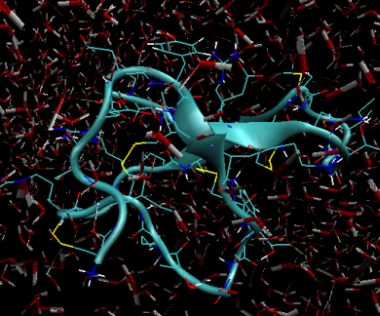
Gromacs data files can be exported to PDB format. The image below shows the final solution state after 20 pico seconds and 25000 iterations. The redwhite "sticks" show the water molecules (2 white hydrogen bonds and 1 red oxygen atom) and immersed in this is the peptide toxin of the funnel web spider, omega-Aga-IVB.
We were expecting the non-openMPI time for one core to exceed the completion time for the openMPI job "distributed" over 1 core and were surprised to see that the times were almost identical. After some head scratching we realized that this was because the non-openMPI job was still a parallel job, using OMP, as the software had been compiled with this option enabled. In fact by not specifying the -nt parameter the job automatically grabs as many free cores as possible.
Gromacs data files can be exported to PDB format. The image below shows the final solution state after 20 pico seconds and 25000 iterations. The redwhite "sticks" show the water molecules (2 white hydrogen bonds and 1 red oxygen atom) and immersed in this is the peptide toxin of the funnel web spider, omega-Aga-IVB.
Note that the load for the parallel openMPI jobs (the first three jobs) is lower than for the OMP jobs. This is because the load for OMP is kept on one server only, while openMPI distributes the load over as many servers as the job parameters allow. The trade off however is that the network communication between the distributed processes is high. Despite this the completion times between OpenMPI and OMP were fairly similar:
We were expecting the non-openMPI time for one core to exceed the completion time for the openMPI job "distributed" over 1 core and were surprised to see that the times were almost identical. After some head scratching we realized that this was because the non-openMPI job was still a parallel job, using OMP, as the software had been compiled with this option enabled. In fact by not specifying the -nt parameter the job automatically grabs as many free cores as possible.
Gromacs data files can be exported to PDB format. The image below shows the final solution state after 20 pico seconds and 25000 iterations. The redwhite "sticks" show the water molecules (2 white hydrogen bonds and 1 red oxygen atom) and immersed in this is the peptide toxin of the funnel web spider, omega-Aga-IVB.