
Using the UCT HPC cluster.
Our head node is called hpc.uct.ac.za as well as hex.uct.ac.za. When using these guides we refer to hpc.uct.ac.za, however you can substitute hex for hpc if you wish.
We acknowledge that a Linux cluster can be intimidating for first time users. This page serves to describe the physical layout of the cluster and how you will interact with it. We strongly recommend reading this entire page before running your first job. Please email one of the administrators if something is not clear to you. As you are given direct access to the operating system running work directly on the head node degrades the performance of the scheduler and negatively impacts other users. Running jobs directly on the head node will not be tolerated and will lead to the immediate suspension of your account in order to protect the scheduler.
Architecture: Any HPC cluster in general has a head node (a Linux server/computer) that you login to. There are additional Linux servers called worker nodes which are very powerful machines with many CPUs (we call them cores) and lots of RAM (memory). These are grouped into partitions according to their characteristics; number of cores, RAM, GPUs etc. There are also two large file systems (hard drives) for storing your data which are network-attached to all the servers.
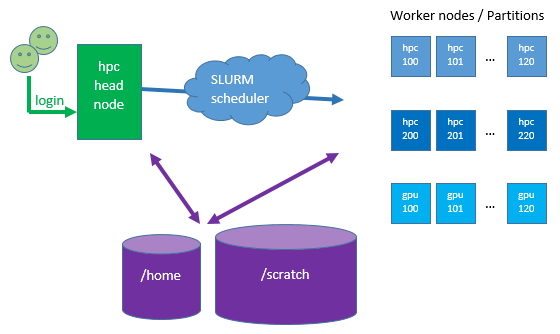
You will not run jobs (or any command that would induce load) on the head node. Instead, you will create sbatch (shell) scripts and submit these to the scheduler. The scheduler, (SLURM – Simple Linux Utility for Resource Management) will queue your job until there is space on the worker nodes for it to run. This is to ensure that your job uses the CPUs that are assigned to it and does not clash with other researchers’ jobs which would cause contention on the CPUs making all the jobs run slowly.
The storage system or disks are shared between the head node and all worker nodes, hence any data changed on the disk of a worker node is immediately visible on the head node. The ‘disks’ or storage areas that you have access to are called /home and /scratch. They are similar to the C: or D: drives on your computer and you can copy data between them or upload\download data to or from them. If you upload large amounts of data to the cluster you should upload it directly to /scratch, you do not have to “transfer” it via your home volume. /scratch is just another disk.
Apart from the data that your job will write to disk the scheduler will also create an output file named slurm-123456.out, where 123456 is your jobID or job number. Output that your software would normally write to the screen is saved to this file, so viewing this file with the cat or tail commands can show you how well your job is progressing.
If you want you can ask the scheduler to email you when your job starts and\or finishes. Once your job is submitted there are commands you can run which will allow you to check its status or cancel it.
And most importantly you can submit many jobs at once, not just one at a time, although there is no guarantee that they will all run simultaneously, that is the task of the scheduler to ensure fairness on the queue that you share with other researchers.
If you are not familiar with Linux and are new to using clusters then everything below this section is important. Please take the time to read through it.
It is not possible to describe all use cases, however our most commonly asked questions deal with:
If this is your first time accessing a Linux server you will need to download and install a SSH client. We have a separate help page describing how to do this as well as how to transfer files to and from the cluster
If you are not familiar with linux you may find the default editor Vi intimidating. We have installed two other text editors; Nano is similar to Windows notepad and gedit almost identical but it does require you to run an X client on your desktop.
The ICTS High Performance Cluster uses SLURM to schedule jobs. There is one head node that researchers login to in order to submit jobs. The /home and /scratch partitions on the head node are mounted on all worker nodes, regardless of series. Resources are assigned to partitions which can be thought of as queues.
| Partition | Description | Nodes | Cores / node | Max cores / user | Time limit |
|---|---|---|---|---|---|
| ada : 100 series | Fast cores, less RAM | 100-115 | 48 | 200 | 250 hours |
| ada : 200 series | Slower cores, more RAM | 200-226 | 40 | 200 | 250 hours |
| curie | Alternate partition | 600-607 | 64 | 64 | 100 hours |
| l40s | GPU partition | 012-015 | 48 (4 GPU cards) | 96 (8 GPU cards) | 48 hours |
| gpumk | Compsci GPU partition | 005-008 | 32 (4 GPU cards) | varies | Private |
| a100 | GPU partition | 009-011 | 56 (4 GPU cards) | varies | varies |
| sadacc | Private | 128-135 | 44 (4 GPU cards) | 176 | 1 hour |
Researchers are assigned to an account which is analogous to a group, normally their department or research group, for instance maths, compsci etc. A researcher may also be assigned to additional accounts. Accounts may also be limited to specific partitions, hence a researcher may submit to the ada partition using their maths account, but may only submit to the GPU partition using their mathsgpu account for example.
If you do not specify a time limit then your job will inherit the default maximum partition time. You may however not want this, as the shorter the wall time specified, the more likely a queued (waiting) job is to be selected by the backfill scheduler to jump the queue. The scheduler can only do this if the job guarantees it will finish in a specified, short period of time.
Initial job node limitations
Parallel jobs using MPI can address cores on multiple nodes using the nodes= directive. However if your job is not capable of running in MPI mode reserving more than one node will not make your job run faster. As some researchers do not fully understand the distinction between nodes and cores we sometimes find non-MPI jobs reserving cores on nodes that are not used. In order to avoid this waste of resource we have set the number of nodes that can be reserved for a job to 1. In order to increase this limit please contact us.
Undergraduate and Honours work
The UCT HPC cluster is intended for postgraduate supervised work that will result in a MSc, PhD or peer reviewed paper published in an accredited journal. We do however grant undergrads limited access to the cluster as we believe that this is the best time to learn how to use Linux and HPC schedulers. Undergrads are limited to 80 simultaneous cores and 100 hour jobs and honours students are limited to 120 simultaneous cores 170 hour jobs.
Time format in SLURM:
Before starting it is important to understand the format of the time parameter to avoid ambiguity and confusion. Acceptable time formats include “minutes”, “minutes:seconds”, “hours:minutes:seconds”, “days-hours”, “days-hours:minutes” and “days-hours:minutes:seconds”. This option applies to job and step allocations. If you do not specify a wall time then the partition’s default wall time will be applied to your job. This will potentially disadvantage you in terms of job priority, so if you know that your job will finish in a certain time then specify that wall time in your sbatch script. When you do this the scheduler can more easily backfill your job, in other words allow it to jump the queue and start sooner.
Some examples:
50 = 50 minutes
50:00 = 50 minutes
50:00:00 = 50 hours
2-2 = 50 hours (2 days and 2 hours)
2-2:00 = 50 hours (2 days and 2 hours)
2-2:00:00 = 50 hours (2 days and 2 hours)
Software Selection
The cluster has many software packages installed in /opt/exp_soft, including duplicate versions. In order to self-provision a software package you will make use of the pre-installed Environment Modules. The module command allows one to view available software packages, ‘activate’ a package, list which packages you have activated and ‘de-activate’ a package. Some modules auto-activate other supporting packages.
The cluster software page has detailed instructions on how to make use of the module command and also has a list of currently installed software. Please check the list before asking us to install software.
Some software packages (R and Python for example) have additional libraries that can be installed. We normally make the base versions of these packages available and additional libraries can be self-installed. We follow this approach because installing additional libraries into the base packages can lead to dependency version conflicts. However we do also install modified versions of base packages into which we will install additional libraries if possible, so check with us first before you start down the self-service approach. For example python/miniconda3-py39 is the base version of Python 3.95 while python/miniconda3-qiime2 and python/miniconda3-py39-picrust2 are dedicated to Python with qiime and picrust respectively and we will install anything into python/miniconda3-py39-usr up until the point we reach a dependency conflict. This also helps to cut down on duplicate Python and miniconda installs in /home.
Submitting jobs
Basic jobs:
Create a shell script with parameters similar to the one below:
#!/bin/sh #SBATCH --account maths #SBATCH --partition=ada #SBATCH --time=10:00:00 #SBATCH --nodes=1 --ntasks=4 #SBATCH --job-name="MyMathsJob" #SBATCH --mail-user=MyEmail@uct.ac.za #SBATCH --mail-type=ALL /opt/exp_soft/softwareX/xyz -o /home/fred/testA15/myfile.txt
There are some example scripts at the bottom of this page…
There must be no spaces between the start of the line and #SBATCH. There must be no spaces between the # and SBATCH.
All the #SBATCH directives must be at the top of the file with no other commands mixed in between them.
Your account will most likely be different to maths. Please use quotes to encapsulate your job name.
Always include the nodes= directive, even if you only want 1 node. If you ask for 10 cores and don’t’ specify nodes you may get 2 cores on one node and 8 on another. Not very useful for non-MPI jobs.
You can then submit the job by typing: sbatch myscript.sh
NB: Do not run: bash myscript.sh as this just runs the script on the head node. The command to launch the job is sbatch myscript.sh.
The directive “nodes” is the number of worker nodes or servers required and “ntasks” is the total number of cores per job. Do not request more than 1 node unless you know that your software is capable of parallel-distributed computation. If you specify nodes > 1 then at least one thread will be assigned to each of the additional servers. A thread will be spawned for each core\ntasks that you request. If your job is not capable of parallel processing then the only reason you would request ntasks > 1 is to claim more RAM\memory, see Memory control below. While we are happy to investigate and help you to understand your software it is your responsibility as a researcher to know how these tools work.
You do not need to cd to the directory from which the job is launched.
If you wish to run more than one job at a time in the same folder you must ensure that each job’s output is directed to a different file, otherwise data files will conflict or overwrite one another. In the above example the second job’s output should be directed to myfile2.txt.
While the job runs on the worker node standard output and standard error (the screen output you’d see if you ran on a desktop) is written to a .out file. If the screen output of your software fills up the disk your job will fail. It is best to ensure that your job output is directed to a file in /home or /scratch, possibly with a command line argument or the linux redirect > function. In addition it is recommended that you disable all spurious or unnecessary program output to minimize on disk space usage, particularly for long job runs.
Memory control:
Like the CPU cores memory is a limited resource. The mem-per-cpu directive allows you to specify how much RAM is needed. If your job exceeds the maximum RAM/core your job will be terminated. The amount of RAM per node can be found on the cluster architecture page. There exists a ratio of RAM per core on each server. For the current ada nodes which have 40 cores and 384GB of RAM the ratio is 1:9. If your job reserves 2 cores it has access to 18GB of RAM. If your job requires 40GB of RAM you will need to reserve this by either using the mem-per-core directive or by reserving 5 cores. It is your responsibility to determine how much RAM your jobs require.
Parallel jobs:
Parallel jobs write to one file system regardless of which worker node they start on. However this does mean that each job a user submits is required to start in a unique folder if the software that the job runs is not capable of specifying unique data files.
As an example, user fred has a home directory /home/fred/ on the head node, and this directory is also mounted on each worker node. This means that if fred created /home/fred/myfile.txt on the head node, this file is also immediately present on each worker node. Fred now submits a job. The job initially lands on hpc102. Slurm now uses OpenMPI to start parallel versions of this job on nodes hpc101 and hpc103 for example. Each of the three nodes writes data to /home/fred/myfile.txt
If fred now submits another job and the software that fred is using cannot distinguish between concurrently running versions then data written to /home/fred/myfile.txt will be intermingled and/or corrupted. Hence it is critical that non-concurrent capable software be launched from unique directories. If fred wants to run 3 concurrent jobs then the following need to be created: /home/fred/job1, /home/fred/job2 and /home/fred/job3. The shell script that controls the job must have a change directory command in it in order to select the correct directory.
The cluster uses OpenMPI to control parallel jobs. To launch a parallel aware program one generally uses mpirun, however as SLURM is tightly coupled with OpenMPI there are some distinctions to launching mpi jobs manually; one does not need to specify a hostfile\machinefile nor does one need to specify the number of threads in the command line. SLURM has its own wrapper to mpirun, srun. Also be aware that unlike Torque\PBS there is no symmetrical geometry, if you request 2 nodes and 4 cores then SLURM will do the bare minimum to satisfy your request by running 3 threads on one node and 1 thread on the second. To retain symmetry use –ntasks-per-node=X where X is the number of threads per node you wish to use. It is critical that the shell script specifies how many servers (nodes) and cores will be reserved. This will inhibit other user’s jobs from trying to run on the same cores which would cause contention, slowing down both jobs. Use the #SBATCH directives to specify the nodes and cores.
It is also important to understand that asking for more cores will not necessarily make your job run faster, it depends entirely on the nature of the algorithm in your code. In addition, splitting a job over multiple nodes when it could be satisfied with the cores on one node may slow your job down slightly due to inter-node communication and data aggregation. In other words 8 cores on 1 node is in general better than 4 cores on 2 nodes. Only use multiple nodes if you have to.
#!/bin/sh #SBATCH --account maths #SBATCH --partition=ada #SBATCH --time=10:00:00 #SBATCH --nodes=2 --ntasks=8 --ntasks-per-node=4 #SBATCH --job-name="MyMathsJob" #SBATCH --mail-user=MyEmail@uct.ac.za #SBATCH --mail-type=ALL module load mpi/openmpi-4.0.1 srun /home/fred/mympiprog
This shell script tells SLURM that 2 nodes and a total of exactly 4 cores on each node should be reserved. Note that if –ntasks-per-node was not specified then the first node would have used 7 cores and the second node would have used 1 core. Unless specified the scheduler will not distribute the threads symmetrically. Mpirun is coupled to the scheduler and it is not necessary to specify a host file.
Please note that if your code uses OMP or PTHREADS we have set OMP_NUM_THREADS=1 on all worker nodes by default as some researchers launch OMP jobs without setting this variable which results in the code grabbing all cores on the worker node. You are welcome to override this if needed with:
export OMP_NUM_THREADS=$SLURM_NTASKS
Or if running in hybrid node with an OMP job distributed via MPI:
export OMP_NUM_THREADS=$SLURM_TASKS_PER_NODE
If your code is capable of running in parallel but does not use OpenMPI and requires a command line argument for the number of cores or threads such as -n 30 or -t 30 then you can link the reserved cores to this with the $SLURM_NTASKS variable for example -n $SLURM_NTASKS instead of -n 30, for example:
#!/bin/sh
#SBATCH --account maths
#SBATCH --partition=ada
#SBATCH --time=10:00:00
#SBATCH --nodes=1 --ntasks=30
#SBATCH --job-name="MyMathsJob"
#SBATCH --mail-user=MyEmail@uct.ac.za
#SBATCH --mail-type=ALL
myprog -np ${SLURM_NTASKS} -in data.txt
Aside: while the curly brackets around the variable name above are not strictly needed in this case, some software require no spaces between the switch arguments and the number, for example -t16. In this case you would need -t${SLURM_NTASKS}
If your software requires a host file or machine file, in other words a text file with the names of all the worker nodes that have been reserved, you can run MakeHostFile > machinefile
salloc:
The salloc command is used to interactively allocate a SLURM job allocation. When salloc successfully obtains the requested allocation, it then runs the command specified by the user. Finally, when the user specified command is complete, salloc relinquishes the job allocation. Entering the following at the head node returns a confirmation and prompt once resources are available:
salloc --account maths --partition=ada --time=10:00:00 --nodes=1 --ntasks=1 salloc: Granted job allocation 2060 bob@srvcnthpc001:~$>
User bob is still logged into the head node but can now use srun to issue commands which will run on the assigned resources, even though the prompt still indicates the head node.
srvcnthpc001 ~$ srun cat /etc/hostname srvcnthpc101.uct.ac.za
Typing exit relinquishes the resources and ends the job.
bob@srvcnthpc001:~$ exit exit salloc: Relinquishing job allocation 2060 salloc: Job allocation 2060 has been revoked. bob@srvcnthpc001:~$
It is possible to launch a cluster job directly from the command line (or a script).
srun -A maths --partition=ada --time=1000:00 --nodes=1 --ntasks=1 /home/fred/myprog -o /home/fred/out.txt
The prompt is frozen until the job completes.
Interactive Jobs:
An interactive job gives you command line access to a worker node. From the head node type:
sintx
The cluster will indicate that you are starting an interactive job and your prompt will change to that of a worker node. In addition the hostname format changes to black on white text:
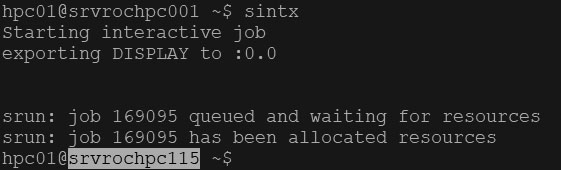
Now any command you type is executed on that node. If you do not see the text “Starting interactive job” then you are still on the head node and should not run any heavy load processes.
Unlike salloc your commands do not need to be prefaced with srun unless you are running OpenMPI code.
Type exit to end the job and you will return to the head node.
You can specify additional cluster parameters with the sintx command just as in an sbatch file:
sintx --ntasks=20 --account=maths --partition=ada
Account and partition parameters are not mandatory unless you have access to more than one partition.
In addition sintx automatically creates a DISPLAY environment variable should you wish to export a graphical display back to your workstation. You will however need to be running an Xclient on your desktop.
Advanced node selection:
The ada partition consists of several tranches of worker nodes purchased over time. The characteristics of these nodes are uniform within their tranches, however the 100 series are more powerful than the 200 series. The architecture of these nodes can be viewed here. By default your jobs will target the 100 range in the ada partition. When this fills up jobs will spill over into the 200 series. If you want to avoid this and rather have your job queue until a faster node is available you must use the following directive:
#SBATCH --constraint=large
The 200 series have a slightly better RAM\core ratio, if you want your jobs to target these nodes then specify the following directive:
#SBATCH --constraint=small
Type squeue to see a list of running jobs
andy@srvcnthpc001:~$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2143 testl MyBatchJob andy PD 0:00 1 (resources)
2144 test MPImemjobA fred R 2:25:02 2 hpc106,107
2150 test MPImemjobB fred R 1:15:27 2 hpc108,109
Here user andy wants to see why his job is not running, it’s most likely that user fred is consuming all the available resources. Note that in SLURM worker nodes can belong to multiple partitions with different queuing attributes and it is possible that some researcher’s jobs may overtake other queued jobs. This is often the case where a research group has paid for the servers and therefore has priority on them.
We also provide an alternate command, qstat, if you only wish to see the status of your own jobs.
To cancel a job type scancel jobid
NB, in the examples below, myaccount refers to your job submission account (medicine, maths or eleceng for example), not your user account.
Recommended sbatch script for submitting R jobs:
#!/bin/sh #SBATCH --account=myaccount #SBATCH --partition=ada #SBATCH --nodes=1 --ntasks=1 #SBATCH --time=10:00 #SBATCH --job-name="MyJob" module load software/R-4.3.3 R CMD BATCH MyRScript.R
Recommended sbatch script for submitting Python Jobs:
#!/bin/sh #SBATCH --account=myaccount #SBATCH --partition=ada #SBATCH --nodes=1 --ntasks=1 #SBATCH --time=10:00 #SBATCH --job-name="MyJob" module load python/miniconda3-py3.12 python MyPythonScript.py
We make use of Miniconda to deploy Python. The version of Python is appended to the module name, in this case it is version 3.12.
Recommended sbatch script for submitting MATLAB jobs:
#!/bin/sh #SBATCH --account=myaccount #SBATCH --partition=ada #SBATCH --nodes=1 --ntasks=1 #SBATCH --time=10:00 #SBATCH --job-name="MyJob" module load software/matlab-R2024b matlab -batch MyMatlabScript
NB, it is essential to add the -batch parameter and also exclude the .m extension of the MATLAB script.